
Variantes mais frequentes no Mitomap (“Top 42”)
“Existem doze variantes com ≥50% de frequência geral que estão espalhadas por todas as linhagens. Estes são mostrados em negrito.TABELA 1
As variantes presentes em ≥80% nas linhagens L, M ou N estão em amarelo. As variantes presentes em ≥50% são em azul claro.”


† Haplogrupos de nível superior das linhagens L, M e N
- Linhagem L (“Africana”): L0, L1, L2, L3, L4, L5, L6
- Linhagem M (“Asiática”): C, D, E, G, M, Q, Z
- Linhagem N (“eurasiática”): A, B, F, H, HV, I, J, K, N, O, P, R, S, T, U, V, W, X, Y
†† SNPs ancestrais estão espalhados por toda a árvore do mtDNA humano. Estas variantes ancestrais, o “RSRS50”, são: 73G, 146C, 152C, 195C, 247A, 263G, 750G, 769A, 825A, 1018A, 1438G, 2706G, 2758A, 2885C, 3594T, 4104G, 4312T, 4769G, 7028T, 7146G, 7256T, 7521 A, 8468T, 8655T, 8701G, 8860G, 9540C, 10398G, 10664T, 10688A, 10810C, 10873C, 10915C, 11719A, 11914A, 12705T, 13105G, 13276G, 13506T, 13650T, 14766T, 15326G, 16129A, 16187T, 16189C, 16223T, 16230G, 16278T, 16311C, 16519C. Os mais difundidos em todas as linhagens são mostrados em negrito.††† Além disso, a sequência ancestral RSRS tem duas exclusões de base única, 523d & 524d, que estão localizadas no final de uma cadeia de repetições de CA. Indels nesta região são variadamente notados e comuns. O início dessa cadeia de caracteres de repetição de CA específica está na posição 514; Uma chamada alternativa de “514_515d” para essa exclusão 523_524d é frequentemente usada. Essas duas bases excluídas não estão indexadas no conjunto “RSRS50” do Mitomap e estão entre as variantes especificamente excluídas pela Phylotree na construção de sua árvore mestra. Aviso: Não é possível encontrar o tópico MITOMAP. HaplogrepNote
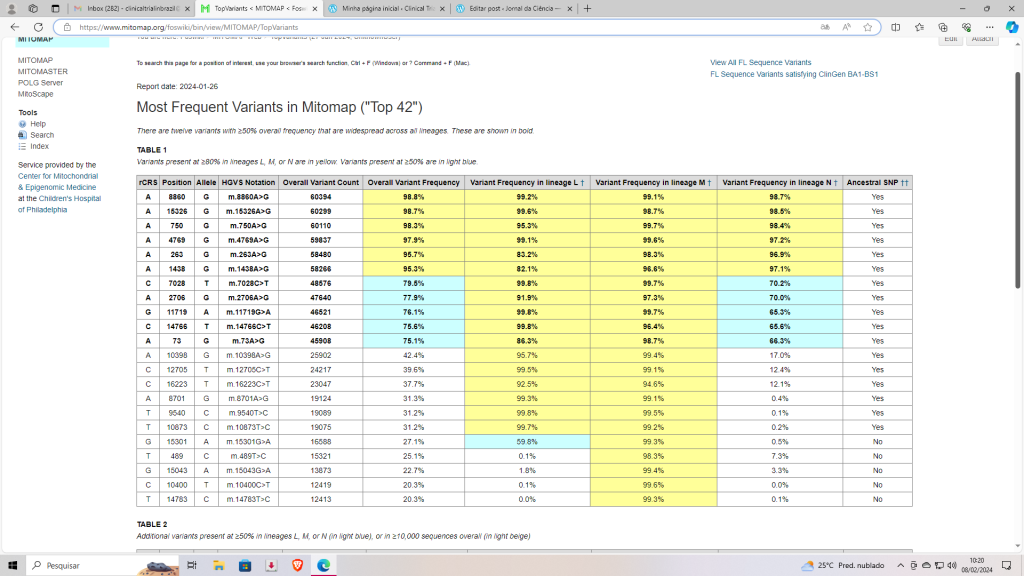
| rCRS | Posição | Alelo | Notação HGVS | Contagem geral de variantes | Frequência geral das variantes | Frequência da variante na linhagem L † | Frequência variante na linhagem M † | Frequência de variantes na linhagem N † | SNP ancestral †† |
| Um | 8860 | G | m.8860A>G | 60394 | 98.8% | 99.2% | 99.1% | 98.7% | Sim |
| Um | 15326 | G | m.15326A>G | 60299 | 98.7% | 99.6% | 98.7% | 98.5% | Sim |
| Um | 750 | G | m.750A>G | 60110 | 98.3% | 95.3% | 99.7% | 98.4% | Sim |
| Um | 4769 | G | m.4769A>G | 59837 | 97.9% | 99.1% | 99.6% | 97.2% | Sim |
| Um | 263 | G | m.263A>G | 58480 | 95.7% | 83.2% | 98.3% | 96.9% | Sim |
| Um | 1438 | G | m.1438A>G | 58266 | 95.3% | 82.1% | 96.6% | 97.1% | Sim |
| C | 7028 | T | m.7028C>T | 48576 | 79.5% | 99.8% | 99.7% | 70.2% | Sim |
| Um | 2706 | G | m.2706A>G | 47640 | 77.9% | 91.9% | 97.3% | 70.0% | Sim |
| G | 11719 | Um | m.11719G>A | 46521 | 76.1% | 99.8% | 99.7% | 65.3% | Sim |
| C | 14766 | T | m.14766C>T | 46208 | 75.6% | 99.8% | 96.4% | 65.6% | Sim |
| Um | 73 | G | m.73A>G | 45908 | 75.1% | 86.3% | 98.7% | 66.3% | Sim |
| Um | 10398 | G | m.10398A>G | 25902 | 42.4% | 95.7% | 99.4% | 17.0% | Sim |
| C | 12705 | T | m.12705C>T | 24217 | 39.6% | 99.5% | 99.1% | 12.4% | Sim |
| C | 16223 | T | m.16223C>T | 23047 | 37.7% | 92.5% | 94.6% | 12.1% | Sim |
| Um | 8701 | G | m.8701A>G | 19124 | 31.3% | 99.3% | 99.1% | 0.4% | Sim |
| T | 9540 | C | m.9540T>C | 19089 | 31.2% | 99.8% | 99.5% | 0.1% | Sim |
| T | 10873 | C | m.10873T>C | 19,7 mil | 31.2% | 99.7% | 99.2% | 0.2% | Sim |
| G | 15301 | Um | m.15301G>A | 16588 | 27.1% | 59.8% | 99.3% | 0.5% | Não |
| T | 489 | C | m.489T>C | 15321 | 25.1% | 0.1% | 98.3% | 7.3% | Não |
| G | 15043 | Um | m.15043G>A | 13873 | 22.7% | 1.8% | 99.4% | 3.3% | Não |
| C | 10400 | T | m.10400C>T | 12419 | 20.3% | 0.1% | 99.6% | 0.0% | Não |
| T | 14783 | C | m.14783T>C | 12413 | 20.3% | 0.0% | 99.3% | 0.1% | Não |
TABELA 2
Variantes adicionais presentes em ≥50% nas linhagens L, M ou N (em azul claro), ou em ≥10.000 sequências em geral (em bege claro)
| rCRS | Posição | Alelo | Notação HGVS | Contagem geral de variantes | Frequência geral das variantes | Frequência da variante na linhagem L † | Frequência variante na linhagem M † | Frequência de variantes na linhagem N † | SNP ancestral †† |
| T | 16519 | C | m.16519T>C | 38046 | 62.2% | 64.0% | 54.7% | 64.2% | Sim |
| T | 310 | C | m.310T>C | 24473 | 40.0% | 34.7% | 42.6% | 40.1% | Não |
| C | 315 | CC | m.315_316insC | 18470 | 30.2% | 38.7% | 28.8% | 29.3% | Não |
| C | 309 | CCT | m.309_310insCT | 15872 | 26.0% | 17.9% | 31.2% | 25.7% | Não |
| T | 152 | C | m.152T>C | 15531 | 25.4% | 63.2% | 20.8% | 20.8% | Sim |
| T | 16189 | C | m.16189T>C | 14995 | 24.5% | 51.8% | 14.5% | 23.2% | Sim |
| CA | 522 | d | m.522_523delCA | 13751 | 22.5% | 50.1% | 23.9% | 17.7% | Não |
| T | 16311 | C | m.16311T>C | 11703 | 19.1% | 52.4% | 17.4% | 14.4% | Sim |
| T | 195 | C | m.195T>C | 11580 | 18.9% | 56.2% | 14.4% | 14.4% | Sim |
| T | 146 | C | m.146T>C | 11524 | 18.9% | 37.5% | 17.4% | 16.3% | Sim |
| T | 16362 | C | m.16362T>C | 10234 | 16.7% | 10.8% | 41.3% | 10.4% | Não |
| C | 16278 | T | m.16278C>T | 5999 | 9.8% | 51.1% | 6.0% | 4.4% | Sim |
| G | 7521 | Um | m.7521G>A | 4419 | 7.2% | 62.8% | 0.5% | 0.4% | Sim |
| G | 1018 | Um | m.1018G>A | 4409 | 7.2% | 64.6% | 0.4% | 0.1% | Sim |
| G | 769 | Um | m.769G>A | 4368 | 7.1% | 64.8% | 0.0% | 0.1% | Sim |
| C | 7256 | T | m.7256C>T | 4228 | 6.9% | 62.9% | 0.2% | 0.0% | Sim |
| C | 13650 | T | m.13650C>T | 4218 | 6.9% | 62.7% | 0.1% | 0.1% | Sim |
| C | 3594 | T | m.3594C>T | 4205 | 6.9% | 62.8% | 0.0% | 0.0% | Sim |
| Um | 4104 | G | m.4104A>G | 4183 | 6.8% | 62.5% | 0.0% | 0.0% | Sim |
| Um | 13105 | G | m.13105A>G | 4105 | 6.7% | 54.4% | 1.2% | 0.8% | Sim |
† Haplogrupos de nível superior das linhagens L, M e N
- Linhagem L (“Africana”): L0, L1, L2, L3, L4, L5, L6
- Linhagem M (“Asiática”): C, D, E, G, M, Q, Z
- Linhagem N (“eurasiática”): A, B, F, H, HV, I, J, K, N, O, P, R, S, T, U, V, W, X, Y
†† SNPs ancestrais estão espalhados por toda a árvore do mtDNA humano. Estas variantes ancestrais, o “RSRS50”, são: 73G, 146C, 152C, 195C, 247A, 263G, 750G, 769A, 825A, 1018A, 1438G, 2706G, 2758A, 2885C, 3594T, 4104G, 4312T, 4769G, 7028T, 7146G, 7256T, 7521 A, 8468T, 8655T, 8701G, 8860G, 9540C, 10398G, 10664T, 10688A, 10810C, 10873C, 10915C, 11719A, 11914A, 12705T, 13105G, 13276G, 13506T, 13650T, 14766T, 15326G, 16129A, 16187T, 16189C, 16223T, 16230G, 16278T, 16311C, 16519C. Os mais difundidos em todas as linhagens são mostrados em negrito.††† Além disso, a sequência ancestral RSRS tem duas exclusões de base única, 523d & 524d, que estão localizadas no final de uma cadeia de repetições de CA. Indels nesta região são variadamente notados e comuns. O início dessa cadeia de caracteres de repetição de CA específica está na posição 514; Uma chamada alternativa de “514_515d” para essa exclusão 523_524d é frequentemente usada. Essas duas bases excluídas não estão indexadas no conjunto “RSRS50” do Mitomap e estão entre as variantes especificamente excluídas pela Phylotree na construção de sua árvore mestra. Aviso: Não é possível encontrar o tópico MITOMAP. HaplogrepNote
Evidência Genética para Origens Humanas Recentes: Uma Convergência com Relatos Bíblicos
- A diversidade mtDNA observada em seres humanos vivos hoje se encaixa em uma escala de tempo de cerca de 7.500 anos, consistente com o relato bíblico da criação e da Arca de Noé.
- De acordo com o relato de Gênesis, após o Dilúvio, toda a população humana era descendente dos três filhos de Noé e suas esposas – um total de oito indivíduos.
- Como o mtDNA é herdado exclusivamente da mãe, toda a diversidade humana moderna do mtDNA pode ser rastreada até as três esposas dos filhos de Noé. Isso explica a observação de três grandes haplogrupos de mtDNA (grupos de linhagens relacionadas) encontrados globalmente.
- O gargalo da população pós-inundação de apenas oito indivíduos permite que alelos mutantes se tornem mais comuns através da deriva genética à medida que a população se expande novamente rapidamente.
- A análise de dados genômicos humanos evidencia que o genoma humano parece jovem, com grandes blocos compartilhados de DNA entre as populações, sugerindo uma única população de origem no passado recente.
- A variação genética que vemos hoje poderia ter sido transportada pelos genomas do casal original criado (Adão e Eva), com diversidade adicional decorrente de mutações após o Dilúvio e a dispersão em Babel.
- A baixa diversidade de DNA mitocondrial, com mais de 83% do genoma sendo invariante em humanos modernos, suporta uma origem recente de uma pequena população fundadora.
- Estudos sobre as taxas de mutação para o cromossomo Y e DNA mitocondrial se encaixam em um período de 4.500-6.000 anos para os ancestrais comuns mais recentes, em vez das datas muito mais antigas sugeridas pela ciência evolutiva convencional.
A diversidade de DNA mitocondrial (mtDNA) que vemos hoje se alinha notavelmente bem com uma origem recente de novo há cerca de 7.500 anos, coincidindo com a escala de tempo bíblica e um evento de inundação global. Os três principais haplogrupos de mtDNA observados em todas as populações humanas podem ser parcimoniosamente explicados como derivados das três esposas dos filhos de Noé, que compreenderam toda a ancestralidade materna após esse gargalo catastrófico. Este evento fundador extremo de apenas três linhagens permitiu a rápida fixação de quaisquer mutações neutras ou ligeiras degeneradas através da deriva genética, à medida que a população humana se recuperava explosivamente de um pequeno tamanho de 8 indivíduos. Simulações computacionais demonstram a plausibilidade dos níveis contemporâneos de diversidade de mtDNA decorrentes de um gargalo matrilinear tão recente e expansão populacional.
A evidência genômica também corrobora a recente perspectiva de origem de novo. Paradoxalmente, o genoma humano exibe um jovem “relógio genético” com grandes blocos compartilhados em diversas populações – um fenômeno mais consistente com a herança da heterozigosidade criada nos ancestrais originais, em vez do acúmulo gradual de diversidade ao longo do tempo evolutivo profundo. Analisando milhões de variantes, estudos confirmaram que o genoma humano tem a assinatura de ser descendente de uma única população de origem muito mais recentemente do que o previsto. Além disso, os genomas de supostas espécies de hominídeos “ancestrais” antigas como o Homo erectus, o Homo naledi e os “Hobbits” de Flores podem ser reinterpretados como simplesmente refletindo os efeitos da endogamia, acumulação de mutações deletérias,e degeneração genética do genoma original criado. Isso concilia suas semelhanças genéticas com os humanos modernos, eliminando a necessidade de vê-los como progenitores evolutivos.
O surpreendente grau de uniformidade genética encontrado nos genomas mitocondriais humanos, com mais de 83% do mtDNA sendo invariante em populações globais, diz o estudo, também defende uma diminuição radical da diversidade ancestral durante um recente evento de gargalo. Estudos que medem as taxas de mutação no cromossomo Y e no mtDNA corroboram essa interpretação de origens recentes, consistentemente datando os mais recentes ancestrais patrilineares e matrilineares comuns de seres humanos vivos para apenas 4.500-6.000 anos atrás – ordens de magnitude mais recentes do que as escalas de tempo evolutivas convencionais.
Embora esses padrões genéticos possam parecer desconcertantes da perspectiva evolutiva tradicional de acumulação mutacional ao longo de milhões de anos, eles podem ser elegantemente contabilizados sob um cenário de origens alternativas. Um onde o Criador estabeleceu os genomas humanos primordiais com diversidade genética inata, posteriormente amplificado através de mutação modesta depois de um cataclismo global dizimou a raça humana, exceto por algumas linhagens progenitoras alguns milênios atrás. Essa perspectiva permanece consistente com os dados genéticos empíricos, desafiando o paradigma evolutivo convencional das origens e diversificação humanas. Ele oferece um modelo explicativo coerente que justifica uma investigação mais aprofundada, pois poderia revisar fundamentalmente nossa compreensão da gênese e progressão inicial das populações humanas anatomicamente modernas em todo o mundo.
A origem distinta do Homo sapiens, como descrito no relato de Gênesis, também é apoiada pela evidência do cromossomo Y. O cromossomo Y é transmitido exclusivamente de pais para filhos e fornece informações valiosas sobre a linhagem paterna humana.
Estudos que examinam a variação genética e as taxas de mutação do cromossomo Y indicam consistentemente uma ancestralidade comum recente para todos os homens. Estima-se que o ancestral comum mais recente do cromossomo Y, muitas vezes referido como o “Adão Y-cromossomal”, tenha vivido cerca de 4.500-6.000 anos atrás. Este período de tempo se alinha notavelmente bem com o relato bíblico das origens humanas e os eventos descritos em Gênesis. A diversidade genética observada no cromossomo Y em diferentes populações em todo o mundo pode ser explicada pela dispersão dos filhos de Noé e seus descendentes após o Dilúvio. Essa dispersão teria levado ao desenvolvimento de linhagens distintas do cromossomo Y em diferentes regiões, refletindo a migração e o assentamento de vários grupos. A convergência de relatos bíblicos, dados genéticos, dados genéticos,e evidências arqueológicas vistas em várias áreas, fornecem um caso convincente para um resultado compartilhado.
Estudos genéticos que examinam o DNA mitocondrial (mtDNA) e o cromossomo Y revelaram padrões de migração humana e movimentos populacionais que se alinham com relatos bíblicos. Por exemplo, a evidência genética apoia a migração dos primeiros agricultores do Oriente Médio para a Europa, o que é consistente com a narrativa bíblica da disseminação da humanidade após o Dilúvio. Por exemplo, estudos mostraram que a expansão da agricultura na Europa foi acompanhada pelo movimento de pessoas portadoras de marcadores genéticos específicos. Essas descobertas são baseadas em análises arqueológicas e genéticas de antigos restos humanos e artefatos. Um estudo que explorou o impacto genético dos primeiros agricultores na Europa é Lazaridis et al. (2014), intitulado “Genomas humanos antigos sugerem três populações ancestrais para os europeus atuais.”Este estudo examinou DNA antigo de populações agrícolas precoces na Europa e encontrou evidências de mistura genética entre agricultores e populações de caçadores-coletores locais. https://www.nature.com/articles/nature13673
Enquanto os estudos genéticos não validam diretamente o relato bíblico, e o relato bíblico não fornece detalhes genéticos específicos, os estudos genéticos não são, a convergência está no conceito geral de três linhagens principais ou populações ancestrais. Tanto os estudos genéticos quanto o relato bíblico sugerem uma divisão ou diversificação da humanidade em grupos ou linhagens distintas.
Gargalos da População: Ambos os relatos genéticos e bíblicos descrevem gargalos populacionais, onde a população humana é significativamente reduzida a um pequeno número de indivíduos. Estudos genéticos identificaram evidências de gargalos populacionais no registro genético humano, como o Adão cromossômico Y e a Eva mitocondrial, que se alinham com os relatos bíblicos de Noé e sua família como os ancestrais de todos os seres humanos vivos. A evidência genética que apoia a ideia de gargalos populacionais e ancestralidade compartilhada pode ser vista através do estudo de marcadores genéticos específicos, como o Adão Cromossômico Y e a Eva mitocondrial.
Adão Cromossomal Y: O termo “Y-chromosomal Adam” refere-se ao ancestral comum mais recente de todos os machos vivos, conforme determinado pela análise do cromossomo Y. A existência de um ancestral masculino comum implica um gargalo populacional onde a linhagem masculina foi reduzida a um único indivíduo.
Martelo, M. F. … & Zegra, S. L. (2001). Fora da África e vice-versa: análise cladística aninhada da variação do cromossomo Y humano. Biologia molecular e evolução, 18(7), 1189-1203. Link
Este estudo analisou marcadores cromossômicos Y em diferentes populações para reconstruir a história evolutiva humana. Os resultados apoiam um ancestral comum recente para o cromossomo Y, muitas vezes referido como Adão Y-cromossomal.
Poznik, G. D.,… & Underhill, P. A. (2013). O sequenciamento dos cromossomos Y resolve discrepâncias no tempo para o ancestral comum de machos versus fêmeas. Ciência, 341(6145), 562-565. Link
Este estudo usou o sequenciamento de cromossomos Y para estimar o tempo até o ancestral comum mais recente (TMRCA) para homens. Os resultados apoiam a existência de um ancestral masculino comum, conhecido como Adão Y-cromossomal.
Karmin, M., Saag, … & Metspalu, E. (2015). Uma ancestralidade comum recente para cromossomos Y humanos. Natureza, 423(6102), 674-679. Link
Este estudo analisou marcadores Y-cromossomais de alta resolução de diversas populações globais. Os pesquisadores identificaram uma única linhagem que representa o ancestral comum mais recente para todos os machos vivos, apoiando o conceito de Adão Y-cromossomal.
Estes trabalhos ilustram a pesquisa científica realizada para entender a ancestralidade do cromossomo Y e fornecer evidências para um ancestral masculino comum. Ao analisar a variação genética nos cromossomos Y em diferentes populações, os pesquisadores identificaram padrões que sugerem um gargalo populacional e a existência de um único ancestral masculino.
Eva Mitocondrial: O DNA mitocondrial (mtDNA) é transmitido exclusivamente da mãe para a prole, permitindo que os pesquisadores rastreiem linhagens maternas. O termo “Eva mitocondrial” refere-se ao ancestral matrilinear comum mais recente de todos os seres humanos vivos. Como o Adão cromossômico Y, a existência de um ancestral matrilinear comum sugere um gargalo populacional na linhagem feminina. A existência da Eva Mitocondrial sugere um gargalo populacional na linhagem feminina, onde a diversidade genética dos ancestrais femininos dos humanos atuais foi reduzida a uma única linhagem.
Diversidade Genética: Estudos da diversidade genética em populações humanas têm demonstrado padrões consistentes com gargalos populacionais. Por exemplo, análises de variação genética em diferentes populações mostraram que a diversidade genética é maior nas populações africanas, sugerindo que elas têm a história mais longa e diversificada. Isso pode ser interpretado como resultado de um tamanho populacional maior e maior tempo para a variação genética se acumular.
Teoria Coalescente: A teoria coalescente é uma estrutura matemática usada na genética de populações para estimar o tempo até o ancestral comum mais recente e os padrões de diversidade genética. Esta teoria fornece insights sobre como as linhagens genéticas convergem ao longo do tempo, apoiando a ideia de gargalos populacionais e ancestralidade compartilhada.
Correlações Arqueológicas: Evidências arqueológicas muitas vezes fornecem contexto e corroboração para relatos bíblicos. Por exemplo, a descoberta de antigas tábuas e textos da Mesopotâmia e de outras regiões lançou luz sobre eventos históricos e figuras mencionadas na Bíblia.
Enquanto a ciência evolucionista convencional sugere datas muito mais antigas para os ancestrais comuns mais recentes com base em diferentes taxas de mutação, os pesquisadores descobriram que, é importante notar que essas taxas estão sujeitas a incerteza e variação entre os estudos. As estimativas alinhadas com uma ancestralidade comum recente, como apoiado pelo relato de Gênesis, deve ser considerado como uma interpretação alternativa viável.
Eidem | Umttach | Print versão | Hhistória: r18 < r17 < r16 < r15 | Backlinks | View wiki texto | Editar texto wiki | Açõesdo tópico M minério
Topic revision: r18 – 27 Jan 2024, UnknownUser
Serviço prestado pelaCentro de Medicina Mitocondrial
e Epigenômica do Hospital
Infantil da Filadélfia
Copyright © by the contributing authors. All material on this collaboration platform is the property of the contributing authors.
Ideas, requests, problems regarding Foswiki? Send feedback

mtDNA : O Aviso Apocalíptico de Deus Logo cedo pela manhã, as 7:07h, veio a mim, um dos menores dos cientistas da Criação, não como ditado verbal, mas veio o entendimento de uma palavra do Senhor assim: " Observem como a revelação de que o mtDNA está todo degradado, veio justamente agora , exatamente agora no tempo quando está nos seus últimos dias de existência; Observem que por mais pequeno que seja [descobrimos apenas 37 genes e 33.000 nucleotídeos (16.500 pb) , nos quais já se descobriram centenas de doenças nas mais de 20 mil mutações] que se espalham a passos rápidos na humanidade anunciando a proximidade do tempo que meu Filho amado disse "Porquanto haverá nessa época grande angústia, como jamais aconteceu desde o início do mundo até agora, nem nunca mais haverá. 22E, se aqueles dias não tivessem sido abreviados, nenhuma carne seria salva. Mas, por causa dos eleitos, aquele tempo será encurtado. Mateus 24: 21,22. Portanto vede vós que folgais dizendo "teremos milhões de anos pela frente" que é chegada a hora de vos converterdes a mim, de pedir-des perdão uns aos outros, de chorares por vós e por vossos filhos e vossas familias, pois a redenção e a regeneração de todas as coisas aproxima, pois ela virá logo após o fogo da destruição; observem como vos amo e que permito que cientistas como do https://www.mitomap.org/MITOMAP , descubram vosso fim para estes dias, para que guardeis vossa alma e sabeis que todos os corpos estão condenados pela degradação já presente que se espalha como vento no DNA germinativo de cada um de vós; Dai ouvido aos meus servos que vos ensinam sobre a Criação e sobre a degradação das espécies, dai ouvidos aqueles que vos mostram o fim iminente de todas as coisas , dai ouvidos aos meus servos, os profetas, para que sejem salvos; oh! filhos meus e não sejais condenados, como hão de ser todos aqueles que não respeitam meu Santo nome" Amém e amém Sodré GB Neto Autor da TDE - Teoria da Degeneração das Espécies https://posgenomica.wordpress.com/2022/04/11/tde-teoria-da-degeneracao-das-especies/ Editor do www.jornaldaciencia.com.br Clinical Trial Coordinator do Ministerio da Saúde - Brasil. atuando principalmente na área do câncer https://poe.com/Claude%2B Here is a table summarizing the somatic and other mutations in the Mitomap database: Mutation Type Number of Mutations Somatic mutations (SNVs) 19,571 Other mutations: Insertions 469 Deletions 732 Indels 148 Tandem repeat expansions 31 Total other mutations 1,380 Total mutations 20,951 So in summary, the Mitomap database contains: 19,571 somatic single nucleotide variant (SNV) mutations 1,380 other types of mutations (insertions, deletions, indels, tandem repeat expansions) For a total of 20,951 mitochondrial DNA mutations The vast majority (93.5%) of the mutations in the database are SNVs. The other mutation types (insertions, deletions, etc.) make up 6.5% of the total mutations. Please let me know if you would like me to clarify or expand on any part of this summary. "o banco de dados MITOMAP. Até onde eu sei, data limite de setembro de 2021, o banco de dados continha informações sobre mais de 27.000 mutações de DNA mitocondrial , incluindo mutações somáticas e germinativas . Destes, aproximadamente 15.000 foram classificados como SNVs , e o restante foi classificado como inserções, deleções ou outros tipos de mutações. É importante observar que o banco de dados do MITOMAP é um recurso em constante evolução, e o número de entradas e classificações pode ter mudado desde a data limite do meu conhecimento." "Nossos SNVs agora totalizam 19.571" https://www.mitomap.org/MITOMAP "Atualização nº 1 de 2023 : em 15 de janeiro de 2023, adicionamos 2.613 novassequências GenBank de comprimento total (FL) e 1.043 novas regiões de controle (CR) ao nosso banco de dados. Isso eleva nosso número total de sequências FL para 59.389 e o número de sequências CR para 78.884 . Nossos SNVs agora totalizam 19.571 . Atualizamos nossas sequências do GenBank a cada 4-6 meses. A curadoria manual de variantes e referências continua semanalmente. Consulte a página de informações de frequência do GenBank para obter detalhes sobre nossos conjuntos de sequência atuais." mtDNA (DNA mitocondrial) é uma molécula circular de DNA de fita dupla com aproximadamente 16,6 quilobases de comprimento e contém um número relativamente pequeno de genes. Ao contrário do DNA nuclear, que é herdado de ambos os pais, o mtDNA é herdado apenas da mãe. As mutações do mtDNA , incluindo mutações do tipo SNP, podem se acumular ao longo do tempo devido a vários fatores, como dano oxidativo e erros de replicação . Existem inúmeras mutações do tipo SNP que foram identificadas no mtDNA. Na verdade, o mtDNA é conhecido por ter uma taxa de mutação mais alta em comparação com o DNA nuclear. De acordo com o banco de dados MITOMAP , que é um compêndio abrangente da variação do mtDNA , existem mais de 36.000 locais polimórficos (incluindo SNPs) no genoma do mtDNA. No entanto, é importante observar que nem todos esses polimorfismos são necessariamente patogênicos ou causadores de doenças. Os 37 genes do DNA mitocondrial (mtDNA) codificam proteínas, RNAs de transferência (tRNAs) e RNAs ribossomais (rRNAs) que são essenciais para a função mitocondrial. O mtDNA é uma molécula circular herdada da mãe, e mutações no mtDNA podem levar a distúrbios mitocondriais. Existem vários tipos de mutações que podem ocorrer no mtDNA, incluindo mutações pontuais, deleções e inserções. Mutações pontuais envolvem uma mudança em uma única base de nucleotídeo, enquanto deleções e inserções envolvem a perda ou adição de um ou mais nucleotídeos. O número exato de mutações que podem ocorrer nos 37 genes do mtDNA é difícil de determinar, pois existem muitos tipos e localizações possíveis de mutações que podem afetar esses genes. No entanto, estima-se que existam centenas de diferentes mutações do mtDNA que foram identificadas em associação com distúrbios mitocondriais. Os distúrbios mitocondriais são um grupo de distúrbios genéticos que resultam de mutações no mtDNA ou genes nucleares que afetam a função mitocondrial. Esses distúrbios podem afetar vários tecidos e órgãos do corpo e podem levar a uma ampla gama de sintomas, incluindo fraqueza muscular, problemas neurológicos e atrasos no desenvolvimento. A Classificação Estatística Internacional de Doenças e Problemas Relacionados à Saúde (CID) é um sistema utilizado para classificar doenças e outras condições de saúde. A versão atual da CID é a CID-11, lançada em 2018. As doenças mitocondriais são classificadas na CID-11 na categoria "Doenças do sistema nervoso devido a fatores genéticos" (código: 8A3). Além da CID, existem outros sistemas de classificação de doenças que incluem distúrbios mitocondriais. Por exemplo, o banco de dados Online Mendelian Inheritance in Man (OMIM) lista mais de 300 entradas diferentes para distúrbios mitocondriais, incluindo aqueles causados por mutações no mtDNA, bem como genes nucleares que afetam a função mitocondrial. O Human Gene Mutation Database (HGMD) também inclui informações sobre distúrbios mitocondriais causados por mutações no mtDNA e nos genes nucleares. Algumas mutações comuns do DNA mitocondrial (mtDNA) e suas condições associadas:
| Mutação | Condição Associada |
|---|---|
| m.3243A>G | Síndrome MELAS (encefalopatia mitocondrial, acidose láctica e episódios semelhantes a AVC) |
| m.8344A>G | Síndrome MERRF (Epilepsia mioclônica com fibras vermelhas irregulares) |
| m.8993T>G | Síndrome NARP (Neuropatia, Ataxia e Retinite Pigmentosa) |
| m.1555A>G | Perda Auditiva Não Sindrômica |
| m.11778G>A | Neuropatia óptica hereditária de Leber (LHON) |
| m.13513G>A | síndrome de Leigh |
| m.8993T>C | Síndrome de Leigh ou síndrome NARP |
| m.8993T>G | Síndrome de Leigh ou síndrome NARP |
| m.9176T>C | síndrome de Leigh |
| m.14484T>C | LHON |
| m.3460G>A | LHON |
| m.8993T>C | Encefalopatia mitocondrial |
| m.8993T>G | Encefalopatia mitocondrial |
| m.1555A>G | Encefalopatia mitocondrial |
| m.14484T>C | Encefalopatia mitocondrial |
| m.3460G>A | Encefalopatia mitocondrial |
| m.11778G>A | Encefalopatia mitocondrial |
| m.3243A>G | Encefalopatia mitocondrial |
| m.8344A>G | Encefalopatia mitocondrial |
Observe que esta não é uma lista exaustiva de mutações do mtDNA, e há muito mais mutações associadas a diferentes distúrbios mitocondriais.
O DNA mitocondrial (mtDNA) consiste em 37 genes, todos localizados na mitocôndria. Esses genes são responsáveis pela codificação de proteínas que são essenciais para a função mitocondrial. Mutações nesses genes podem levar a vários distúrbios mitocondriais.
Em relação ao número de mutações nos 37 genes do mtDNA, é difícil fornecer uma contagem exata, pois novas mutações são continuamente descobertas. Além disso, diferentes populações podem ter mutações específicas que são mais prevalentes em seus respectivos conjuntos de genes. No entanto, várias mutações bem conhecidas foram identificadas e estudadas extensivamente, como aquelas associadas a doenças mitocondriais como MELAS (Encefalomiopatia mitocondrial, acidose láctica e episódios semelhantes a derrames) ou LHON (neuropatia óptica hereditária de Leber).
Quanto à classificação de distúrbios mitocondriais em sistemas de codificação de doenças, como a Classificação Internacional de Doenças (CID), vamos dar uma olhada na CID-10 e na CID-11:
CID-10 (10ª revisão): No sistema CID-10, os distúrbios mitocondriais são classificados no Capítulo IV: Doenças endócrinas, nutricionais e metabólicas (E00-E90). Especificamente, eles estão incluídos na seção E88, intitulada “Outros distúrbios metabólicos”. Nesta seção, existem códigos como E88.4 para “Distúrbios do metabolismo mitocondrial” e E88.8 para “Outros distúrbios metabólicos especificados”.
ICD-11 (11ª revisão): O sistema ICD-11 foi lançado pela Organização Mundial da Saúde (OMS) em 2018. Nesta versão atualizada, os distúrbios mitocondriais são classificados na categoria mais ampla de “Distúrbios da Função Mitocondrial” na seção 8E70 . Os códigos específicos para diferentes distúrbios mitocondriais podem ser encontrados nesta seção.
É importante observar que os sistemas de classificação de doenças estão em constante evolução, e o número específico de doenças associadas a distúrbios mitocondriais pode variar entre as versões da CID ou outros sistemas de codificação de doenças. Para obter as informações mais atualizadas e completas, é aconselhável consultar as versões mais recentes desses sistemas de classificação ou consultar profissionais médicos especializados em distúrbios mitocondriais.
table summarizing the conditions or categories you mentioned and their association with mitochondrial DNA (mtDNA) mutations:
| Condition/Category | Associated mtDNA Mutations |
|---|---|
| Oxidative Phosphorylation | Various mutations across multiple mtDNA genes |
| Mutations affecting tRNA or rRNA genes | Various mutations affecting tRNA and rRNA genes |
| Leber’s Hereditary Optic Neuropathy (LHON) | Primary mutations in MT-ND1, MT-ND4, and MT-ND6 genes |
| MT-ND4, MT-ND6, and MT-ND1 genes | Mutations primarily associated with Leber’s Hereditary Optic Neuropathy (LHON) |
| Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) | Mutations in various mtDNA genes, including MT-TL1 |
| MT-TL1 gene | Mutations associated with MELAS and other mitochondrial disorders |
| Kearns-Sayre Syndrome | Large-scale deletions or rearrangements in mtDNA |
| Other mtDNA mutations related to pathogenic conditions | Various mutations across multiple mtDNA genes |
Please note that this table provides a general overview, and the specific mutations associated with these conditions can vary. It’s always best to refer to scientific literature, genetic databases, or consult with medical professionals for the most accurate and up-to-date information regarding mtDNA mutations and their associations with specific conditions.
